By our Scientific Director Dr Ann Smith
Haemoglobinopathies – the facts and treatment options
Sickle cell anaemia and thalassaemia belong to a group of conditions called the haemoglobinopathies. These inherited conditions involve problems with haemoglobin (Hb) which is an iron containing protein that is found in red blood cells and is responsible for transporting oxygen from the lungs to the rest of the body. (1, 2)
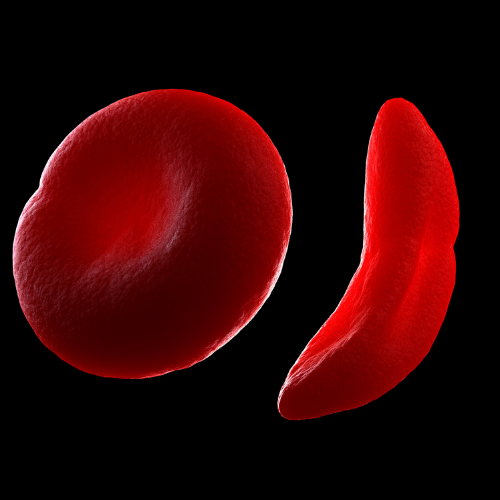
How does haemoblobin work?
Each Hb molecule is made up of four sub units with two alpha globin and two beta globin sub units. The globin units are sometimes called chains and carry heme molecules that in turn contain iron.
Each iron rich heme molecule can bind onto one oxygen molecule which means that each hemoglobin molecule is capable of binding four oxygen molecules. Heme molecules with more oxygen, for example in red blood cells that have just picked up oxygen in the lungs, are brighter red. Because of this, oxygenated blood in the arterial system is bright red, while venous blood that is deoxygenated is darker red. Each human red blood cell has 640 million molecules of haemoglobin. (3)
What can go wrong?
Diseases such as sickle cell anaemia and thalassemia decrease the blood’s oxygen-carrying capacity and ability to deliver oxygen to tissues.
Thalassaemia: background
Thalassemia is caused by a defect in either the alpha or the beta subunits of Hb. There are several types of thalassaemia, some relatively mild and others which are very serious. Around 100.000 people are born each year with severe forms of thalassaemia, most usually with Mediterranean, South Asian and African ancestry.
Patients with clinically relevant thalassemia produce a high number of red blood cells, but these cells have lower-than-normal levels of hemoglobin so the oxygen-carrying capacity is reduced. The main types of thalassaemia are alpha and beta. (4, 5, 6, 7)
Alpha Thalassaemia
Alpha thalassaemia, which is common in Southern China, Southeast Asia, India, the Middle East and Africa results when the haemoglobin fails to produce enough alpha globin protein. To make alpha globin protein, four alpha globin genes are required, two from each of our parents. The severity of the disease depends on the how many genes are mutated:
- Alpha thalassaemia minima, sometimes called “silent” or “alpha-thal trait”: one faulty gene producing no symptoms
- Alpha thalassaemia minor: two faulty genes produce mild anaemia
- Haemoglobin H disease: three faulty genes resulting in lifelong anaemia
- Alpha thalassaemia major: four faulty genes which causes the most severe type of thalasaemia. This condition is also known as Bart’s Hydrops Fetalis. As there are no functioning alpha globin genes, this causes a severe life-threatening anaemia in the developing fetus and without intervention, this is incompatible with life following birth.
Beta Thalassemia
Beta thalassaemia tends to occur amongst people with Mediterranean ancestry and is most prevalent in North Africa, West Asia and the Maldives. As with alpha thalassaemia, the severity of the condition depends on the number of genes that are mutated:
- Beta thalassaemia minor: one faulty gene producing no symptoms
- Beta thalassaemia major: results in severe symptoms with the need for frequent blood which can eventually result in buildup of iron in the body, resulting in liver, heart, and hormone problems.
Diagnosis of thalassaemia
In moderate or severe cases, it will typically be apparent that children have thalassemia by the time they are two years old. People who are both carriers with only one gene mutation and no symptoms may not realise that they have a genetic defect until they have children.
Blood and genetic testing are typically undertaken to demonstrate the level of anaemia and to check for faulty genes. It is possible to undertake prenatal testing to show if a developing foetus has thalassemia and can indicate the severity of the condition.
Signs and symptoms of thalassemia (these vary depending on the severity of the condition)
- Jaundice and pallor
- Fatigue
- Chest pain
- Cold hands and feet
- Breathlessness
- Cramps
- Rapid heart beat
- Delayed growth
- Skeletal deformities
- Problems due to iron build up from transfusions i.e. liver and heart damage
Treatments for thalassaemia
- Blood transfusions to help relieve the symptoms of anaemia
- Iron chelation therapy which involves removing excess iron from the blood stream.
- Stem cell transplant may be relevant in severe cases and may offer a cure. The aim is to use chemotherapy to wipe out the impaired blood making bone marrow cells and replace them with donor haematopoietic (blood making) stem cells that can function correctly. Stem cells may be sourced from sibling or matched unrelated donor bone marrow, peripheral blood (following the use of drugs to coax the cells out of the bone marrow into the blood) or from cord blood.
- Gene therapy, the safety and efficacy of which is being investigated in early clinical trials. The technique involves removing some of the patient’s haematopoietic (blood making) stem cells and inserting a normal beta globin gene into the relevant cell type. The genetically engineered stem cells are then re-infused back into the patient following chemotherapy to obliterate the flawed cells. Results to date are encouraging but more work is needed to better understand and refine this treatment (8, 9).
Sickle cell anaemia: background
In sickle cell anaemia, the shape of red blood cells is deformed. Normally red blood cells are flexible biconcave discs, moving easily through blood vessels but in sickle cell anaemia, the cells are elongated and crescent or “sickle” shaped, hence the name of the disorder. This malformation of the red blood cells reduces their ability to carry and deliver oxygen to the tissues, as they cannot effectively pass through narrow blood capillaries.
This blockage, known as sickle crisis can be very painful when it occurs. Sickle cells are fragile and can break up prematurely leading to a shortage of these critical oxygen carrying cells. Sickle cell disease is most common in West Africa and India (10).
Sickle cell anaemia: the genetic defect
Sickle cell anaemia is caused by a mutation in the haemoglobin-beta gene. In individuals with sickle cell disease, abnormal haemoglobin S molecules are produced which causes the red blood cells to become stiff, sticky and sickle shaped – this prevents the (normally smooth) cells from gliding through blood vessels. Children born with sickle cell disease inherit a faulty gene from both parents. If both parents have the genetic defect, there is a 25 percent chance that each child will be born with the disease (11, 12).
Sickle cell carriers: the genetic defect
Carriers have inherited one normal haemoglobin gene and one sickle haemoglobin S gene. Individuals, who carry the sickle cell trait, are relatively symptom free but must be careful in situations where there may be a reduction in the supply of oxygen, for example when having a general anaesthetic or during activities such as deep-sea diving or mountain climbing. Such individuals can pass the defective gene to their offspring (13).
Sickle cell carriers: the genetic defect
Carriers have inherited one normal haemoglobin gene and one sickle haemoglobin S gene. Individuals, who carry the sickle cell trait, are relatively symptom free but must be careful in situations where there may be a reduction in the supply of oxygen, for example when having a general anaesthetic or during activities such as deep-sea diving or mountain climbing. Such individuals can pass the defective gene to their offspring (13).
Diagnosis
Blood testing can detect hemoglobin S, the defective form of hemoglobin. Blood samples from these patients have large numbers of sickled red blood cells – the hallmark of the disease.
Sickle cell disease can also be detected in a developing foetus using amniocentesis, a procedure in which a sample of fluid from around the baby is taken for testing. This can determine whether the foetus has sickle cell disease or carries the sickle cell gene.
Recently, a technique used in conjunction with in vitro fertilization, called pre-implantation genetic diagnosis (PGD) has been developed. This enables parents who carry the sickle cell trait to test embryos for the defective gene before implantation and to choose to implant embryos free of the sickle cell gene.
Signs and symptoms
- Anaemia
- Periodic episodes of pain, resulting from sickle-shaped red blood cells blocking blood flow through small blood capillaries (vaso-occusive crises). The pain, which can be very severe and is a major symptom of sickle cell disease, typically occurs in the chest, abdomen, joints and bones.
- Painful swelling of hands and feet
- Frequent infections
- Delayed growth
- Vision problems
- Stroke
Treatment
Until recently, people with sickle cell disease were not expected to survive childhood. Nowadays due to improved medical care, approximately 50% of patients live to 50 years or more.
- Supportive treatment consists of antibiotics, pain management and blood transfusions.
- Use of an anti-tumour drug called hydroxyurea has been shown to stimulate the production of foetal haemoglobin (HbF), usually found only in newborns. HbF helps prevent the “sickling” of red blood cells.
- Stem cell transplantation is an accepted form of treatment for severe cases of sickle cell disease and offers the only possibility of a cure. The rationale is the same as described in the setting of thalassemia. Chemotherapy is used to destroy the impaired blood making bone marrow cells and replace them with healthy donor stem cells. The stem cells may be sourced from sibling or matched unrelated donor bone marrow, peripheral blood (following the use of drugs to coax the cells out of the bone marrow into the blood) or from umbilical cord blood (14, 15).
- Gene therapy offers promise of a cure. Researchers are considering the possibility of correcting the defective gene in bone marrow from patients. The patients then receive their manipulated cells back following high dose chemotherapy designed to wipe out their defective bone marrow. The aim is to restore the production of normal haemoglobin. This work has currently reached early clinical trial phase but is not yet proven as effective (16).
References
https://www.nature.com/articles/d41586-018-07646-w
https://www.who.int/bulletin/volumes/86/6/06-036673/en/
https://www.ebi.ac.uk/interpro/potm/2005_10/Page1.htm
https://www.ebi.ac.uk/interpro/potm/2005_10/Page2.htm
https://www.nhs.uk/conditions/thalassaemia/
https://ghr.nlm.nih.gov/condition/beta-thalassemia
https://www.nejm.org/doi/full/10.1056/NEJMoa1705342
https://www.bionews.org.uk/page_135491
https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/symptoms-causes/syc-20355876
https://ghr.nlm.nih.gov/condition/sickle-cell-disease#genes
https://www.genome.gov/10001219/learning-about-sickle-cell-disease/
https://www.gov.uk/government/publications/sickle-cell-carrier-description-in-brief
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5451329/pdf/1020976.pdf
Curr Opin Hematol. 2016 Nov; 23(6): 524–529. doi: 10.1097/MOH.0000000000000282
Rate this article: